Le trouble lié à l’utilisation d’opioïdes est considéré comme une maladie chronique récurrente du système nerveux central qui entraîne des troubles de la personnalité, des comorbidités et une mort prématurée. Il se développe à la suite de l’administration à long terme de diverses substances abusives, dont la morphine. L’action pharmacologique de la morphine est associée à la stimulation des récepteurs opioïdes. Les récepteurs opioïdes sont un groupe de récepteurs couplés aux protéines G. L’activation de ces récepteurs par des ligands induit des changements moléculaires significatifs à l’intérieur de la cellule, tels que l’inhibition de l’activité de l’adénylate cyclase, l’activation des canaux potassiques et la réduction de la conductance du calcium. Des données récentes indiquent que d’autres voies de signalisation peuvent également être impliquées dans l’activité de la morphine. Il s’agit notamment de la phospholipase C, des kinases activées par les mitogènes (MAP kinases) ou de la β-arrestine. La présente revue se concentre sur les principaux mécanismes qui sont actuellement considérés comme essentiels dans l’activité et la dépendance à la morphine et qui pourraient faire l’objet d’études plus approfondies.
Listos, J., Łupina, M., Talarek, S., Mazur, A., Orzelska-Górka, J., & Kotlińska, J. (2019). The mechanisms involved in morphine addiction: an overview. International journal of molecular sciences, 20(17), 4302.

Parmi les médicaments opioïdes, la morphine, la codéine, le fentanyl ou la buprénorphine sont considérés comme les analgésiques les plus efficaces pour le traitement des douleurs postopératoires et cancéreuses. Leur administration chronique est associée à un fort potentiel d’abus [1]. En outre, d’autres opioïdes, dont l’héroïne, sont utilisés comme drogues récréatives et ont la capacité d’induire une dépendance aux opioïdes. Le trouble lié à l’utilisation de substances est classé comme une maladie chronique récurrente du système nerveux central (SNC) qui entraîne des troubles de la personnalité, des comorbidités et un décès prématuré [2,3]. Les troubles liés à l’utilisation de substances se développent à la suite d’une administration à long terme de substances présentant un potentiel d’abus et comprennent la dépendance physique et/ou la dépendance psychologique. La dépendance physique est associée à la formation de changements neuroadaptatifs dans le SNC, tant au niveau moléculaire que cellulaire [4,5,6,7,8,9,10,11]. Ces changements sont responsables de l’apparition de signes de sevrage caractéristiques après l’arrêt de la consommation de drogue. Le type et la gravité des signes de sevrage dépendent de divers facteurs, tels que le type de drogue consommée, les doses de drogue, la période de consommation, l’âge du patient, l’âge de la première prise de drogue ou les prédispositions génétiques [12,13]. La dépendance psychologique est définie comme l’utilisation compulsive de drogues pour améliorer la perception du bien-être [14]. Le syndrome typique de la dépendance psychologique chez l’homme comprend un comportement intensifié de recherche de drogue, une capacité de volonté compromise, une prise compulsive de drogue malgré la conscience de ses effets nocifs, ainsi qu’une obsession persistante et récurrente, même après des années d’abstinence.
La recherche scientifique montre qu’en dépit des nombreux projets sociaux, psychologiques et médicaux visant à réduire le phénomène de l’abus de substances, le nombre de personnes souffrant de troubles liés à l’utilisation d’opioïdes ne cesse d’augmenter dans le monde entier. Aujourd’hui, la dépendance aux opioïdes est considérée comme une crise mondiale de santé publique. Selon l’Organisation mondiale de la santé, les décès par surdose d’opioïdes sont passés de 69 000 personnes en 2014 [15] à 118 000 en 2015 [16]. Une augmentation spectaculaire de la consommation d’opioïdes par les mères et du syndrome d’abstinence néonatale a également été observée au cours de la dernière décennie. Par conséquent, en réponse à la crise des opioïdes, les efforts des scientifiques et des gouvernements devraient se concentrer sur plusieurs priorités : améliorer la gestion de la douleur avec des médicaments non addictifs, promouvoir la connaissance des risques liés aux opioïdes ou fournir un soutien à la recherche de pointe sur la douleur et la dépendance. L’objectif de cette revue est de présenter les connaissances actuelles sur les mécanismes impliqués dans l’activité de la morphine qui se développent après son administration aiguë ou chronique. La compréhension des mécanismes de la morphine est importante pour les études ultérieures sur l’activité du système opioïde.
La morphine et d’autres médicaments opioïdes sont capables d’induire un large spectre d’activités pharmacologiques. Au niveau du SNC, ils induisent une forte analgésie, une euphorie, une sédation, une dysrégulation endocrinienne, un myosis, une activité antitussive ou une dépression respiratoire. En outre, ils provoquent des spasmes musculaires et la libération d’histamine dans le système nerveux périphérique. Dans la pratique clinique, il existe de nombreux médicaments opioïdes qui sont principalement utilisés comme analgésiques.
(Tableau 1)
L’action pharmacologique de doses aiguës de morphine est associée à la stimulation des récepteurs opioïdes. Elle interagit principalement avec les récepteurs opioïdes μ. En général, les récepteurs opioïdes peuvent être divisés en sous-types : μ (μ1, μ2, μ3) ; δ (δ1, δ2, δ3) ; et κ (κ1, κ2, κ3) [13,62]. Le nouveau récepteur nociception/orphanine FQ est considéré comme une branche non opioïde de la famille des récepteurs opioïdes [13]. Les récepteurs opioïdes sont un groupe de récepteurs couplés aux protéines G [63]. Ils se composent de sept domaines transmembranaires, de trois boucles extracellulaires et de trois boucles intracellulaires, d’un acide aminé N-terminal extracellulaire et d’un carboxyle C-terminal intracellulaire. Les récepteurs opioïdes sont situés à la fois dans le système nerveux central et périphérique. Les premières données sur la localisation des récepteurs opioïdes dans le système nerveux sont apparues en 1973 [64]. On sait aujourd’hui que les sous-types de récepteurs opioïdes sont situés dans les zones impliquées dans : 1) la transmission de la douleur, comme le thalamus, la moelle rostroventrale (RVM), la zone grise périaqueducale (PAG), le pons ou la corne dorsale de la moelle épinière ; 2) le système de récompense, comme le noyau accumbens, l’aire tegmentale ventrale ou le cortex ; 3) d’autres zones du cerveau, comme l’hypothalamus, l’amygdale, le pallidum ventral, le globus pallidus, le noyau du raphé, l’hippocampe et le bulbe olfactif [64,65,66,67]. Ils sont également présents dans les tissus périphériques, par exemple dans les voies gastro-intestinales et respiratoires [67,68].
La localisation des récepteurs opioïdes dans l’intestin [69] est responsable de la régulation de la motilité et de la sécrétion gastro-intestinales [70]. Par conséquent, les agonistes des récepteurs μ-opioïdes inhibent la vidange gastrique, augmentent le tonus musculaire du pylore et retardent le transit dans l’intestin grêle et le gros intestin. Tous ces effets conduisent à la constipation, l’un des effets indésirables les plus importants de la morphine et des autres médicaments opioïdes.
La présence de récepteurs opioïdes dans les voies respiratoires est associée à d’importantes indications cliniques de la morphine [71]. La morphine est utilisée comme antitussif, également après une intervention chirurgicale sur le système respiratoire, et dans le contrôle de la douleur due au cancer du poumon. Inversement, le surdosage de morphine induit un risque élevé de dépression respiratoire, qui est un facteur limitant important dans le traitement à la morphine [72].
Les récepteurs opioïdes endogènes sont stimulés par des peptides endogènes, tels que les endomorphines, les dynorphines et les enképhalines. Les endomorphines sont constituées de quatre acides aminés, dont deux ligands endogènes (endomorphine-1 et endomorphine-2) qui ont la plus grande affinité et sélectivité pour les récepteurs opioïdes µ dans les systèmes nerveux central et périphérique. Elles sont impliquées dans l’analgésie et la récompense. Les dynorphines (dynorphine A et dynorphine B) exercent leurs effets principalement par l’intermédiaire du récepteur κ-opioïde et ont une affinité moindre pour les récepteurs μ-opioïde et δ-opioïde. Les enképhalines (met-enképhaline et leu-enképhaline) produisent l’effet principalement sur les récepteurs δ, mais elles ont également une affinité pour les récepteurs μ.
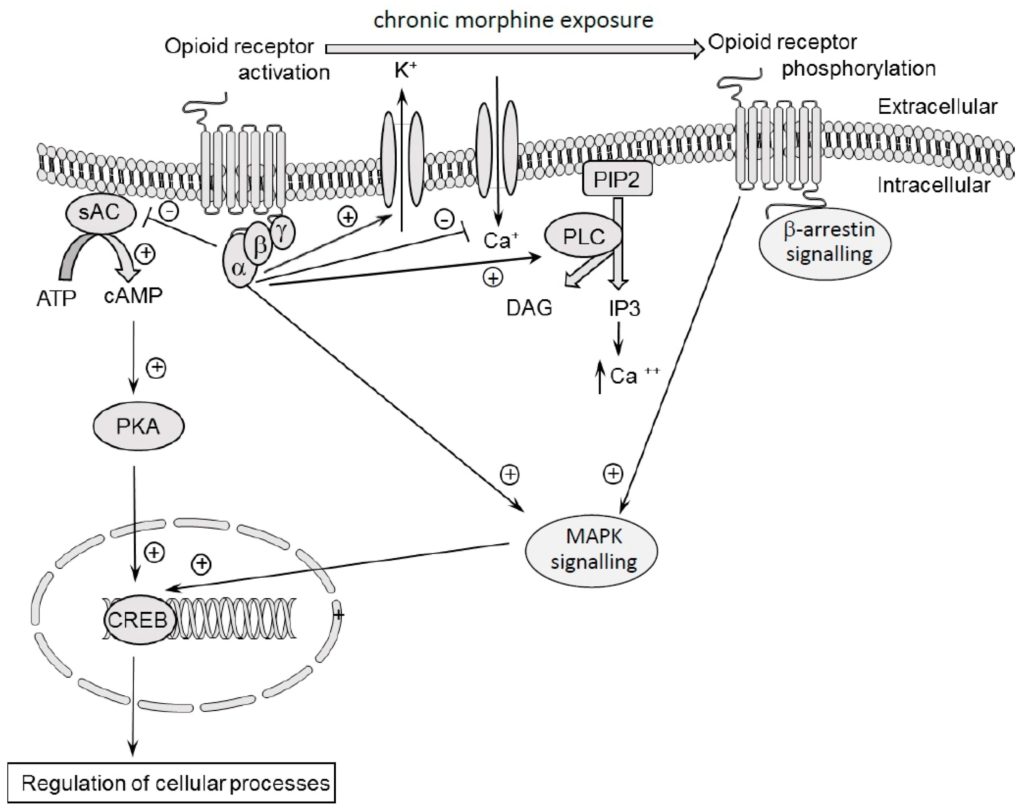
La liaison d’un ligand endogène (molécule d’endomorphine) ou exogène (molécule de morphine) avec un récepteur opioïde entraîne l’activation d’une protéine Go ou Gi et sa phosphorylation par une famille de kinases appelées les récepteurs kinases couplés aux protéines G (GRK). Cela induit des changements moléculaires à l’intérieur de la cellule, notamment la liaison des β-arrestines. La protéine G est composée de trois sous-unités : α, ß et γ. La liaison du ligand au récepteur entraîne l’activation du récepteur opioïde par la liaison du GTP à la sous-unité α, tandis que le complexe α-GTP se dissocie du dimère ßγ-sous-unités. Les deux complexes, α-GTP et dimère ßγ, participent à la transduction du signal intracellulaire. Cela conduit à une inhibition de l’activité de l’adénylate cyclase et à une réduction des niveaux d’adénosine monophosphate cyclique (AMPc) dans la cellule [73,74], ainsi qu’à une suppression de l’activité de la protéine kinase A [75,76]. L’α-GTP active également les voies de la phospholipase-C (PLC) et des protéines kinases activées par des agents mitogènes (MAP) [77]. La PLC hydrolyse le phosphatidylinositol 4,5-bisphosphate (PIP2) en inositol 1,4,5-trisphosphate (IP3) et en diacylglycérol (DAG). L’IP3 augmente la libération de calcium du réticulum endoplasmique, ce qui active la signalisation dépendante du calcium. On observe également l’activation de canaux potassiques (GIRK-3, canal potassique à redressement interne géré par une protéine G) [78], ce qui entraîne une hyperpolarisation accrue de la cellule et, indirectement, une réduction de l’excitabilité cellulaire [77]. Le dimère ßγ bloque directement le canal calcique (canal de type P/Q, de type N et de type L) et réduit la concentration de calcium [79] dans la cellule, ce qui entraîne la suppression d’autres neurotransmetteurs. L’effet de la stimulation des récepteurs opioïdes sur l’activité des canaux potassiques et calciques a été confirmé à plusieurs reprises dans diverses zones du cerveau (hippocampe, noyau du locus coeruleus, région des capsules abdominales, etc.), et ce mécanisme a été considéré comme un effet clé de la stimulation des récepteurs opioïdes [80,81,82,83].
L’exposition chronique à la morphine induit la phosphorylation des récepteurs opioïdes par les GRK. Cette phosphorylation prépare les récepteurs opioïdes à la liaison avec l’arrestine. La liaison de l’arrestine bloque la signalisation médiée par les protéines G, induisant ainsi une désensibilisation des récepteurs opioïdes [84].
Ainsi, les effets pharmacologiques cliniquement importants de la morphine, induits par une seule administration de cette substance, sont liés à des mécanismes moléculaires multidirectionnels qui se produisent à l’intérieur de la cellule. Les effets moléculaires de la morphine sont représentés graphiquement dans la figure 1.
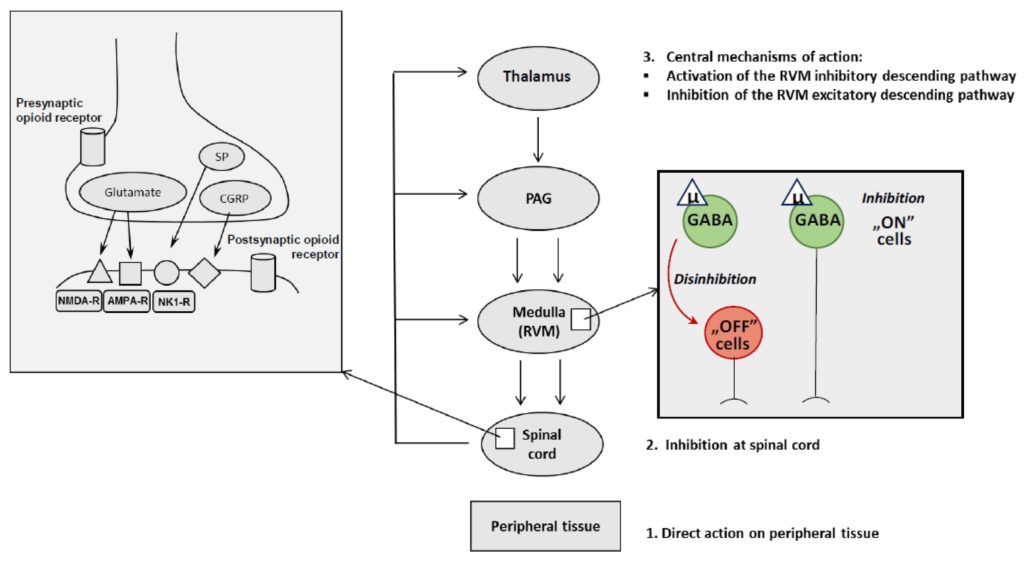
L’analgésie opioïde est fortement associée à l’activation des récepteurs opioïdes μ situés dans le SNC. Ces récepteurs sont localisés dans les régions sous-corticales du cerveau, comme mentionné précédemment, d’où partent les voies descendantes de la douleur, comme dans le thalamus, le PAG et le RVM, ainsi que dans la corne dorsale de la moelle épinière [85]. Au niveau supraspinal, les analgésiques opioïdes stimulent les récepteurs opioïdes μ situés sur les interneurones GABAergiques du RVM, ce qui diminue la libération de GABA. Physiologiquement, le GABA, en agissant sur les récepteurs GABA-A, supprime les cellules « OFF » dans le RVM, ce qui augmente le potentiel d’action. Lorsque le niveau de GABA est réduit, l’inhibition tonique des cellules « OFF » est levée (désinhibition) et le signal des cellules « OFF » supprime la perception de la douleur dans la moelle épinière (régulation descendante de la douleur). En outre, l’activation induite par les opioïdes des récepteurs opioïdes μ sur les cellules GABAergiques « ON » dans le RVM inhibe la mise à feu de ces cellules. Ainsi, la désinhibition des cellules « OFF » et l’inhibition directe des cellules « ON » produisent une analgésie, un effet qui peut être mesuré à l’aide de tests de nociception thermique [86]. En outre, l’amygdale, une zone du cerveau responsable des états émotionnels, peut indirectement modifier la transmission de la douleur.
En ce qui concerne le niveau spinal, les effets analgésiques induits par les opioïdes sont médiés par l’activation des récepteurs opioïdes μ présynaptiques localisés dans la corne dorsale de la moelle épinière. Le déclenchement de ces récepteurs présynaptiques entraîne une hyperpolarisation de la membrane. De tels changements dans la polarisation membranaire conduisent à l’inhibition des médiateurs de la voie de la douleur, tels que le glutamate, la substance P et le peptide lié au gène de la calcitonine (CGRP) à partir des terminaisons des neurones afférents primaires nociceptifs. Par conséquent, la transmission ascendante de la voie de la douleur est atténuée.
Il convient de noter que l’analgésie induite par les opioïdes est un processus complexe dans lequel les récepteurs opioïdes μ peuvent être hétéromérisés avec les récepteurs opioïdes δ ou κ. Les associations hétérodimériques entre récepteurs opioïdes μ-δ, par exemple, peuvent être utilisées comme modèle pour le développement de nouvelles thérapies combinées pour le traitement de la douleur chronique et d’autres pathologies [87]. Les mécanismes de l’analgésie opioïde sont représentés graphiquement dans la figure 2.
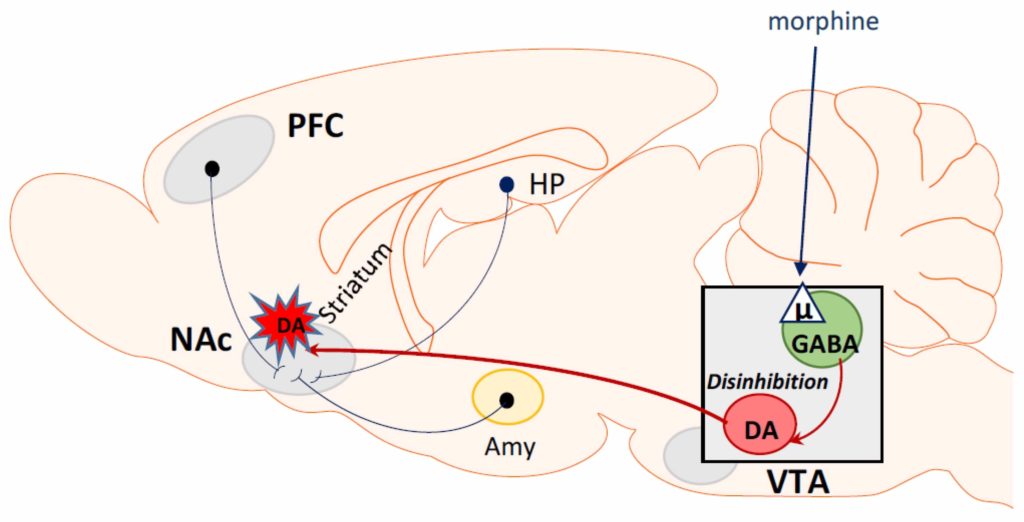
En général, l’effet gratifiant de diverses substances addictives, y compris les opioïdes, est associé à la stimulation des structures du système mésolimbique, telles que l’aire tegmentale ventrale et le noyau accumbens. Cela augmente la libération de dopamine dans le noyau accumbens [88], qui détermine la sensation de plaisir. Cependant, les impulsions provenant d’autres structures cérébrales, telles que le striatum ventral, l’hippocampe, le cortex préfrontal ou l’amygdale, peuvent également stimuler le système mésolimbique [8], affectant les niveaux de dopamine dans le noyau accumbens. Ainsi, une escalade spectaculaire de la consommation de drogues, avec un accès étendu à l’auto-administration de drogues, se caractérise par un dérèglement des voies dopaminergiques gratifiantes dans le cerveau [89,90,91]. Par conséquent, l’effet gratifiant de la morphine et d’autres opioïdes est associé à la stimulation des récepteurs opioïdes μ localisés au niveau des terminaux GABAergiques de l’aire tegmentale ventrale. Cette stimulation inhibe la libération de GABA qui, à son tour, désinhibe les neurones dopaminergiques et entraîne la libération de dopamine dans le noyau accumbens qui induit un sentiment d’euphorie et favorise le développement de la toxicomanie [13,92,93].
Si la dopamine joue un rôle crucial dans l’action gratifiante de la morphine, de nombreux neurotransmetteurs et neuromodulateurs du SNC affectent le système dopaminergique et modulent indirectement divers aspects de la dépendance à la morphine. Ces neurotransmetteurs comprennent le glutamate [94,95], la sérotonine [96], l’acide γ-aminobutyrique (GABA) [97,98], la noradrénaline [96,99], l’adénosine [91], l’oxyde nitrique [100], l’orexine [101], et d’autres encore. La manipulation pharmacologique de ces neurotransmetteurs présents dans la voie de la récompense peut potentiellement modifier l’envie de drogues d’abus. Les mécanismes de l’effet de récompense induit par la morphine sont illustrés graphiquement dans la figure 3.
L’abus chronique de morphine entraîne une dépendance physique et psychologique [8,12,14,102]. La dépendance physique à la morphine se manifeste par des symptômes de sevrage caractéristiques qui peuvent apparaître après un arrêt brutal de l’administration du médicament [103,104]. Les symptômes de sevrage de la morphine chez l’homme comprennent les éternuements, l’écoulement nasal, la toux, les douleurs abdominales, la diarrhée, l’anorexie, l’anxiété et d’autres effets [105,106]. Les effets de sevrage observés chez les animaux comprennent des sauts, des tremblements des pattes, des claquements de dents, des tremblements de chien mouillé et de la diarrhée [103,105,107,108]. Les symptômes de sevrage de la morphine sont évoqués dans des études expérimentales soit par l’arrêt de l’administration chronique de morphine, soit par l’administration d’antagonistes des récepteurs opioïdes. La naloxone, l’antagoniste des récepteurs opioïdes le plus couramment utilisé en pharmacologie expérimentale, est généralement administrée à une dose comprise entre 1 et 6 mg/kg [109,110,111]. La gravité des symptômes de sevrage de la morphine est analysée sur la base du nombre d’épisodes de sevrage [103,111].
(tableau 2)
Certains auteurs suggèrent que la diminution des concentrations de dopamine dans le système mésocorticolimbique joue un rôle critique dans le sevrage de la morphine [112,113,114,115]. Cependant, des neurotransmetteurs tels que la noradrénaline [116], le glutamate [117], la sérotonine [118], l’orexine [119] et le cortisol [120] pourraient également être impliqués dans le sevrage de la morphine. En outre, ces changements dans les neurotransmetteurs s’accompagnent de changements dans les voies de signalisation cellulaire, comme une augmentation significative du niveau d’AMPc [121] et la dérégulation de la voie des MAP kinases (ERK 1/2) [122,123].
La tolérance à la morphine, deuxième paramètre de la dépendance physique à cette substance, se définit comme la nécessité d’augmenter la dose de morphine (déplacement vers la droite de la courbe dose-réponse) pour obtenir le même effet pharmacologique [124]. Le phénomène de tolérance se développe vis-à-vis des effets analgésiques, euphorisants, sédatifs, dépresseurs respiratoires et nauséabonds des opioïdes, mais pas vis-à-vis de leurs effets sur le myosis et la motilité intestinale (constipation) [125]. En général, il existe trois types de tolérance : la tolérance pharmacocinétique, la tolérance apprise et la tolérance pharmacodynamique. La tolérance pharmacocinétique fait référence à des changements dans la distribution ou le métabolisme du médicament, tandis que la tolérance acquise fait référence à une réduction des effets d’un médicament due à des mécanismes compensatoires appris, comme le fait de se comporter normalement tout en restant intoxiqué. La forme la plus importante de tolérance concernant les opioïdes est la tolérance pharmacodynamique. Ce type de tolérance a été lié aux changements neuroadaptatifs qui se produisent après une exposition à long terme au médicament, y compris les changements dans la densité des récepteurs et l’altération du couplage des récepteurs aux protéines G et aux voies de transduction du signal [126].
Au cours des études expérimentales, la tolérance à la morphine est généralement analysée par le biais de la nociception, mesurée par des tests comportementaux, comme le test d’immersion de la queue [103,127] ou le test de la plaque chauffante [128,129] (tableau 2). La tolérance à la morphine peut être modifiée par divers neurotransmetteurs, notamment la dopamine [130,131], mais aussi la sérotonine [132], l’acétylcholine [133], l’orexine [134] ou les endocannabinoïdes [135] et d’autres.
La sensibilisation comportementale est un phénomène impliquant une escalade des réponses comportementales à une exposition répétée à un stimulus tel qu’une drogue d’abus comme la cocaïne ou les opioïdes, après une période sans drogue qui peut être de longue durée, voire de plusieurs années. Cet effet est étroitement lié à l’environnement dans lequel la substance addictive est consommée. La sensibilisation comportementale est un paramètre important pour évaluer le degré de dépendance psychologique. Les études animales reflètent le comportement de recherche de drogue chez l’homme, qui conduit souvent à une rechute de la consommation de drogue [14]. Expérimentalement, la sensibilisation comportementale se manifeste et se mesure généralement par une augmentation de l’activité locomotrice des animaux après l’administration d’une dose provocante de la substance abusée [136,137]. En outre, la sensibilisation comportementale peut également être exprimée par l’augmentation de l’effet gratifiant de la substance addictive. Dans les études animales, ce phénomène peut être observé dans le test de préférence de place conditionnée [100,138,139]. Plus rarement, la sensibilisation peut être observée par l’intensification des signes de sevrage après des périodes de sevrage répétées [91,140] (tableau 2).
Le développement de la sensibilisation comportementale se produit par le biais des changements neuroadaptatifs observés dans la neurotransmission glutamatergique et dopaminergique [102,141,142,143]. La sensibilisation est associée à une augmentation de la libération de dopamine dans les structures mésolimbiques [14,144,145]. Les principales voies impliquées dans la sensibilisation comportementale sont la voie dopaminergique de l’aire tegmentale ventrale et les voies glutamatergiques du cortex préfrontal, qui se terminent toutes deux dans le noyau accumbens.
L’expression de la sensibilisation à la morphine est associée principalement à une augmentation de la libération de dopamine et à des altérations de la sensibilité des récepteurs dopaminergiques D1 dans les structures mésolimbiques, notamment le striatum, le noyau accumbens, l’aire tegmentale ventrale, l’hippocampe et le cortex préfrontal [4,5,6,7,91,146,147]. Chez le rat, le blocage pharmacologique des récepteurs D1 entrave l’expression de la sensibilisation [148]. De même, l’administration d’antagonistes des récepteurs D1 et D2 dans le noyau accumbens inhibe également le développement de la sensibilisation comportementale chez le rat [147]. De plus, des données publiées montrent que l’expression accrue des récepteurs D1 dans l’enveloppe du noyau accumbens, observée au cours du processus de sensibilisation à la morphine, est associée à une activité MAP kinase élevée (ERK 1/2) et que cet effet est réduit par l’antagoniste des récepteurs D1, le SHC 23390 [149]. Par ailleurs, le développement de la sensibilisation comportementale est davantage associé au système glutamatergique et à l’aire tegmentale ventrale, car les antagonistes des récepteurs NMDA et AMPA inhibent l’acquisition, mais pas l’expression, de la sensibilisation comportementale [150,151]. Ainsi, divers changements neuroadaptatifs, y compris des altérations de la densité des récepteurs, du niveau des neurotransmetteurs ou une dérégulation de la signalisation cellulaire, peuvent être responsables de l’expression et de l’acquisition de la sensibilisation comportementale.
Au niveau cellulaire, l’effet principal d’une dose aiguë de morphine est la diminution du niveau de cAMP et l’hyperpolarisation qui est induite par des changements dans l’activité des canaux potassiques et calciques. Cependant, la stimulation chronique des récepteurs opioïdes par la morphine et d’autres ligands opioïdes induit des changements adaptatifs au sein des récepteurs opioïdes. Cela entraîne une diminution de la réponse aiguë des récepteurs. Ces changements sont essentiels pour contrôler l’activité des récepteurs car ils les protègent contre l’hyperstimulation, favorisent la terminaison du signal et régulent leur expression [86]. Par conséquent, la désensibilisation, l’internalisation, la resensibilisation ou la régulation à la baisse des récepteurs opioïdes se développent. Ces mécanismes conduisent à l’atténuation de l’activité pharmacologique de la morphine et d’autres drogues opioïdes, qui est souvent observée après une exposition chronique à ces substances.
En ce qui concerne l’exposition chronique à la morphine, il a été suggéré précédemment que la régulation à la baisse des récepteurs µ était principalement responsable de la réduction de l’activité de la morphine qui se manifestait sous forme de tolérance. Cette hypothèse n’a toutefois pas été confirmée par des expériences, car l’exposition chronique à la morphine n’a pas entraîné de régulation négative des récepteurs μ [152,153]. De même, l’internalisation des récepteurs μ a été précédemment considérée comme un mécanisme neuronal sous-jacent à la tolérance à la morphine. Cependant, les études in vitro ne confirment pas cette hypothèse car la morphine est capable d’induire une forte tolérance mais son effet sur l’internalisation des récepteurs μ est faible [154]. De nos jours, un nombre croissant de preuves relient les mécanismes de tolérance à la désensibilisation des récepteurs opioïdes μ. Il convient de souligner que les définitions de la tolérance cellulaire et de la désensibilisation sont similaires et que, pendant de nombreuses années, ces termes ont été confondus. Tous deux sont définis comme une capacité réduite à répondre à la même dose de drogue. Cependant, la désensibilisation (appelée « tolérance rapide » dans des études antérieures) désigne une réduction progressive de la transduction du signal dans les récepteurs opioïdes induite par l’agoniste, observée dans des modèles in vitro, tandis que la tolérance est observée dans des modèles in vivo. La désensibilisation se développe directement après l’exposition aux opioïdes et s’inverse rapidement en l’absence d’agoniste [84]. La désensibilisation rapide dépend de l’activité des ions potassium et calcium, tandis que la désensibilisation soutenue est liée à l’activité enzymatique (adénylyl cyclase ou MAP kinases). De nos jours, la désensibilisation accrue des récepteurs opioïdes est considérée comme un mécanisme important de la tolérance à la morphine, qui résulte de nombreux changements neuroadaptatifs. La désensibilisation peut être causée par une activité accrue de l’adénylate cyclase et un niveau élevé d’AMPc, par exemple, qui, à son tour, affecte l’activité de la protéine de liaison de l’élément de réponse à l’AMPc (CREB) [155]. En outre, la désensibilisation implique un découplage des protéines G car, chez les animaux traités à la morphine, la liaison du complexe GTPα est réduite par rapport aux animaux témoins. De plus, la désensibilisation des récepteurs µ induite par la morphine est étroitement associée à la dérégulation des niveaux de β-arrestin-1 et de β-arrestin-2 dans la cellule [156,157,158]. La β-arrestine, une protéine cytosolique, est liée à la surface du récepteur opioïde après sa phosphorylation par une classe de sérine/thréonine kinases (GRK). L’activation de la β-arrestine inhibe la signalisation cellulaire, ce qui entraîne directement la désensibilisation du récepteur [86,159]. Le processus de désensibilisation est également produit par une augmentation de la phosphorylation des MAP kinases. Les MAP kinases ont un large éventail de substrats potentiels, y compris des facteurs de transcription contrôlant l’expression des gènes. Le rôle de la kinase régulée par le signal extracellulaire 1/2 (ERK1/2) dans l’effet de l’administration chronique de morphine a également été confirmé, bien que ces résultats soient contradictoires. Narita et al [160] et Macey et al [161] ont observé que l’exposition chronique à la morphine induisait une augmentation de la phosphorylation de ERK1/2, tandis que d’autres auteurs ont montré l’absence d’effet [162,163,164]. Certaines données montrent également le rôle de la phospholipase C dans la désensibilisation induite par la morphine [165]. La phospholipase C augmente le niveau d’autres neurotransmetteurs secondaires, tels que l’inositol- (1,4,5) -triphosphate (IP3) et le 1,2-diacylglycérol (DAG). Ceux-ci entraînent une élévation du taux de calcium dans la cellule. La phospholipase C catalyse également la libération d’acide arachidonique des membranes cellulaires, ce qui participe à la formation de l’inflammation [166]. La phospholipase C joue un rôle important dans l’activité de la morphine. Les inhibiteurs de la phospholipase C potentialisent l’effet antinociceptif d’une dose unique de morphine et réduisent la tolérance à la morphine [167,168].
Les effets moléculaires de la tolérance et de la dépendance à la morphine sont illustrés par la figure 1.
De nouvelles données montrent que l’exposition chronique à des drogues abusives peut induire des interactions épigénétiques complexes au sein d’un génome, régulant ainsi les schémas d’expression des gènes [169,170,171]. Ces modifications épigénétiques comprennent des changements dans la méthylation de l’ADN [172], l’acétylation et la déméthylation des histones, des altérations de l’accessibilité de l’ADN et des modifications de la structure de la chromatine. Elles sont héritées malgré l’absence d’effet sur la structure de l’ADN. Les premières preuves du rôle de la méthylation de l’ADN et de la désacétylation des histones dans l’expression du récepteur opioïde µ ont été publiées en 2007 [173]. Observée dans des cellules de carcinome embryonnaire de souris P19, l’hyperméthylation de l’ADN réduit au silence les gènes des opioïdes µ au niveau transcriptionnel et la déméthylation de l’ADN induit une plus forte expression des gènes des opioïdes µ. En outre, l’expression des récepteurs µ opioïdes a également augmenté après une manipulation pharmacologique, telle que l’administration d’un agent déméthylant (5′-aza-2′-désoxycytidine) et d’inhibiteurs d’histone-désacétylases. Une autre étude a démontré que la modification de la chromatine participe également à l’expression du gène des opioïdes µ [174]. Mashayekhi et al [175] ont montré que les modifications des niveaux d’ARNm du facteur neurotrophique dérivé du cerveau, BDNF, dans l’aire tegmentale ventrale et le locus coeruleus des rats au septième jour d’abstinence de morphine étaient associées à des modifications des histones. Une autre étude a également confirmé le rôle de la méthylation des histones dans les effets de l’exposition chronique à la morphine [176]. Ciccarelli et al [177] ont constaté l’augmentation de l’acétylation des histones pendant le sevrage de la morphine précipité par la naloxone dans l’enveloppe du noyau accumbens et dans le septum latéral. Des expériences ont également montré que l’inhibition pharmacologique de la méthylation de l’ADN par la 5′-aza-2′-désoxycytidine avait une influence sur la préférence de place pour la morphine chez les rats [178,179]. Une étude récente a confirmé l’existence de changements épigénétiques dans de nombreuses structures cérébrales (entre autres : cortex cérébral, cervelet, hippocampe, hypothalamus, bulbe rachidien, etc.) après une exposition aiguë et chronique aux opiacés [180].
Bien que de nombreux progrès aient été réalisés dans la compréhension des mécanismes de l’activité opioïde, on sait peu de choses sur les mécanismes de régulation transcriptionnelle. Il semble qu’ils soient des régulateurs essentiels de l’expression des gènes. Des études complémentaires sont nécessaires pour définir les liens précis entre les altérations épigénétiques et les effets comportementaux de la morphine et d’autres opioïdes.
Étant donné que les drogues morphiniques traditionnelles présentent de nombreux effets indésirables (démangeaisons, constipation, nausées/vomissements, dépression respiratoire ou labilité de l’abus), on a récemment synthétisé des ligands biaisés au niveau du récepteur couplé à la protéine G. Ces composés ont été perçus comme stimulant préférentiellement certaines voies intracellulaires et d’autres. Ces composés ont été perçus comme stimulant préférentiellement certaines voies intracellulaires plutôt que d’autres et produisant moins d’effets secondaires. Ainsi, ces drogues pourraient être plus sûres, plus efficaces et mieux tolérées que la morphine [181]. Jusqu’à présent, plusieurs agonistes des récepteurs opioïdes μ basés sur la protéine G (GPB-MOR) ont été développés [182]. Ils activent préférentiellement la signalisation de la protéine Gαi liée à l’analgésie par rapport à la signalisation de la β-arrestine qui médiatise certains effets indésirables. TRV130 (oliceridine) a été le premier agoniste de ce type à faire l’objet d’un essai clinique [183]. Un autre ligand de GPB-MOR est un composé appelé PZM21, qui représente le premier exemple de découverte basée sur la structure d’un ligand de récepteur couplé à la protéine G biaisé. Le PZM21 s’est révélé prometteur dans les études animales en tant qu’analgésique puissant sans dépression respiratoire et sans effets de renforcement semblables à ceux de la morphine [184]. Cependant, bien que les MOR puissent produire moins de dépression respiratoire et de dysfonctionnement gastro-intestinal à des doses analgésiques que les analgésiques opioïdes actuellement disponibles, ils conservent leur responsabilité en matière d’abus [185]. D’autres candidats thérapeutiques prometteurs pour le soulagement de la douleur et des démangeaisons sont des agonistes du récepteur opioïde κ basés sur la protéine G [186]. L’exemple d’un tel composé est le Triazole. Il n’a pas modifié l’activité locomotrice et n’a pas provoqué de sédation dans les modèles animaux. Ces effets semblent provenir de son incapacité à diminuer la libération de dopamine dans le striatum de la souris, n’affectant donc pas négativement la transmission dopaminergique. En outre, ce composé n’a pas influencé le circuit de la récompense dans le cerveau et n’a donc pas déclenché de signes de dysphorie et d’aversion, contrairement à un agoniste opioïde κ typique. D’une manière générale, les découvertes de nouvelles voies et de nouveaux ligands au niveau des récepteurs opioïdes μ/ κ mettent en évidence les possibilités offertes par les ligands biaisés en tant que nouvelle classe d’analgésiques offrant un soulagement plus efficace et plus sûr de la douleur modérée à sévère.
En résumé, la morphine, agissant sur les récepteurs opioïdes, induit un large spectre d’activité pharmacologique. Cependant, l’administration de morphine à long terme génère une dysrégulation aux niveaux cellulaire et moléculaire dans le cerveau, conduisant à l’addiction. Malgré des études approfondies, la prise en charge efficace des troubles liés aux opioïdes est limitée. Par conséquent, la reconnaissance des mécanismes sous-jacents à la dépendance à la morphine et aux opioïdes semble être extrêmement importante dans la recherche de nouvelles stratégies thérapeutiques pour les consommateurs de morphine et d’opioïdes. La présente étude résume les connaissances actuelles sur l’activité de la morphine et fournit une vue d’ensemble des mécanismes impliqués dans son exposition aiguë et chronique.